

Причины заболевания
Врачи уверены, что основная причина синдрома – это генетический фактор. Но в большинстве случаев специалисты не могут сказать, каким способом передаются гены. Изменений в хромосомном наборе также не выявляют, в некоторых ситуациях наблюдается только раздробление клеток.
Точной причины синдрома Корнелии де Ланге медики не могут назвать, но есть провоцирующие факторы, на фоне которых развиваются патологические процессы:
- Тяжелое инфекционное заболевание, которым переболела женщина в период вынашивания малыша.
- Применение химических препаратов.
- Использование физиотерапевтических процедур.
- Сильная интоксикация организма во время беременности.
- Негативное влияние радиации.
- Заболевания эндокринной системы, которые диагностировали у матери.
- Преклонный возраст родителей.
- Мать и отец являются родственниками.
Ученые больше склоняются к тому, что патологические процессы запускаются наследственными факторами. Однако были зафиксированы ситуации, когда синдром появлялся впервые в роду.
Осложнения и последствия
Ввиду выраженной патологии внутренних органов и сниженного иммунитета, частыми осложнениями являются различные вирусные и инфекционные заболевания. Наличие повышенного внутричерепного давления, может провоцировать судорожный синдром, который проявляется как в виде малых припадков, так и развернутых с клоническими и тоническими судорогами.
В данном случае, последствия этой врожденной патологии, неблагоприятные. Такие дети являются инвалидами и требуют постоянного внимания и ухода. Ввиду нарушений в психическом развитии, социализация в обществе представляется большой проблемой. Как правило, такие дети являются постоянными пациентами психиатрических клиник и интернатов.

Признаки и симптомы
Фенотип CdLS очень разнообразен и описывается как спектр; от Classic CdLS (с большим количеством ключевых функций) до умеренных вариаций с несколькими функциями. У некоторых людей будет небольшое количество функций, но у них нет CdLS.
Ключевая особенность:
- Длинные и / или густые брови
- Короткий нос
- Вогнутый носовой гребень и / или загнутый кверху кончик носа
- Длинный и / или гладкий желобок
- Тонкая киноварь верхней губы и / или опущенные уголки рта
- Отсутствие пальцев рук или ног
- Врожденный диафрагмальная грыжа
Другие характерные особенности:
- Задержка развития и / или Интеллектуальная недееспособность
- Малый дородовой размер и вес при рождении
- Маленький рост
- Микроцефалия (пренатально и / или постнатально)
- Маленькие руки и / или ноги
- Короткий пятый палец
- Гирсутизм
Следующие ниже состояния здоровья чаще встречаются у людей с CdLS, чем у населения в целом.
- Респираторное заболевание
- Сердце дефекты (например, легочный стеноз, VSD, ASD, коарктация аорты )
- Нарушение слуха
- Нарушения зрения (например, птоз, нистагм, высоко миопия, гипертропия )
- Частичное сращение второго и третьего пальцев стопы
- Вогнутые 5-е пальцы (клинодактилия )
- Гастроэзофагеальный рефлюкс
- Желудочно-кишечные аномалии
- Опорно-проблемы
- Сколиоз
- Социальная тревожность
- Судороги
- Волчья пасть
- Проблемы с кормлением
У детей с этим синдромом часто бывают длинные ресницы, густые брови и синофрисы (соединил брови). Волосы на теле могут быть чрезмерными, и больные люди часто короче, чем их ближайшие родственники. У них характерный лицевой фенотип.
 Читайте также:
Читайте также:

Дети с CdLS часто страдают от желудочно-кишечный тракт трудности, особенно гастроэзофагеальный рефлюкс. Рвота, прерывистый плохой аппетит, запор, понос или газообразное вздутие, как известно, является закономерностью в случаях, когда проблемы желудочно-кишечного тракта являются острыми. Симптомы могут варьироваться от легких до тяжелых.
Люди с CdLS могут демонстрировать поведение, которое было описано как «похожее на аутизм», включая самостимуляцию, агрессию, самоповреждение или сильное предпочтение структурированному распорядку дня. Проблемы с поведением в CdLS не являются неизбежными. Многие поведенческие проблемы, связанные с CdLS, являются реактивными (т. Е. Что-то происходит в теле или окружающей среде человека, вызывая такое поведение) и циклическими (приходит и уходит). Часто основная проблема со здоровьем, боль, социальная тревога, стресс из-за окружающей среды или лица, осуществляющего уход, могут быть связаны с изменением поведения. Если причиной является боль или проблема со здоровьем, после лечения поведение уменьшается.
Есть свидетельства некоторых особенностей преждевременного старение включая раннее развитие Пищевод Барретта, остеопороз присутствует в подростковом возрасте, преждевременное поседение волос и некоторые изменения кожи лица, приводящие к более старому внешнему виду по сравнению с хронологическим возрастом.
Список литературы
1. Козлова С.И. Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, Авторская академия; 2007.
2. Барашнев Ю.И., Бахарев В.П., Новиков П.В. Диагностика и лечение врожденных и наследственных заболеваний у детей (путеводитель по клинической генетике). – М.: «Триада-Х»; 2004.
3. Гинтер Е.К. Медицинская генетика. – М.: Медицина; 2003.
4. Жимулев И.Ф. Общая и молекулярная генетика. – Новосибирск: Сиб. унив. изд-во; 2002.
5. Бородулин В.И., Тополянский А.В. Синдромы и симптомы в клинической практике: эпонимический словарьсправочник. – М.: Эксмо; 2009.
6. Berney T.P., Ireland M., Burn J. Behavioral phenotype of Cornelia de Lange syndrome. // Arch. Dis. Child. – 1999. – Vol. 81. – P. 333-336.
-
Читайте также:

7. Deardorff M.A., Krantz I.D. Cornelia de Lange Syndrome // Encyclopedia of Neuroscience. – 2009. – P. 159-162. doi: org/10.1016/b978-008045046-9.01491-1.
8. Badoe E.V. Classical Cornelia de Lange syndrome. // Ghana Medical Journal. – 2010. – Vol. 40, issue 3. – P. 148-150
9. Mundlos S., Horn D. Cornelia de Lange Syndrome. // Limb Malformations. – 2014. – P.215-216. doi: org/10.1007/978-3540-95928-1_85.
10. Белозеров Ю.М. Детская кардиология (наследственные синдромы). – Элиста: ЗАОр «НПП «Джангар»; 2008.
Какая поддержка доступна для семей, пострадавших от Синдром Корнелии де Ланге?
ДКСТ Фонд Великобритании и Ирландии является добровольной организацией, которая является частью сети подобных организаций по всему миру. Он играет важную роль в поддержке родителей, пропаганде научных исследований, а также повышение осведомленности среди специалистов в области здравоохранения, чтобы поощрить раннюю диагностику.
ДКСТ Фонд имеет широкий спектр ресурсов для поддержки семей. Существует телефонная линия помощи работают волонтеры, и он подготовил буклеты и информационные пакеты, дающие важную информацию для семей и специалистов в области здравоохранения. Ежеквартальный бюллетень обсуждает актуальные вопросы, а также содержит новую информацию о последних исследованиях. Семейные встречи проводятся каждые шесть месяцев, чтобы позволить семьям, пострадавшим собираться вместе и обмениваться опытом, и проводится в разных местах, с тем чтобы максимальное посещаемость. Основные конференции также организованы, в том числе международного присутствия, а также есть “мини-конференций” в другое время.
ДКСТ Фонд создал свой собственный научно-консультативный совет (SAC), который существует, чтобы позволить специалистам обмениваться информацией, поддерживать связь друг с другом (например, путем участия в международных конференциях), а также предоставить семьям доступ к достоверной научной информации о ДКСТ.
ДКСТ Фонд также поддерживает медицинские исследования в попытке улучшить диагностику и лечение лиц с ДКСТ, и искать лекарство. Тем не менее, поскольку организация зависит от добровольных пожертвований, средства ограничены.
Естественно, что диагноз ДКСТ оказывает значительное влияние на семьи, а также множество функций и характеристик означает, что каждый человек должен поддерживаться и лечение в индивидуальном порядке. Тем не менее, исследования продолжаются, и организации, такие как Фонд ДКСТ может предложить свою помощь и поддержку как для тех, кто непосредственно затронуты, и специалистам в области здравоохранения, работающих в этой области.
-
Читайте также:

Клиническая картина[править | править код]
- Микроцефалия (уменьшение размеров черепа более чем на 10 % возрастной нормы);
- Брахицефалия (укорочение черепа в сагиттальном направлении, в результате чего поперечный размер головы увеличивается, а продольный уменьшается);
- Тонкие сросшиеся брови; длинные загнутые ресницы; деформированные ушные раковины; маленький нос, открытые вперед ноздри, атрезия хоан; тонкая верхняя губа;
- Микрогения; высокое нёбо или расщелина нёба; нарушение прорезывания зубов;
- Миопия, косоглазие, астигматизм, атрофия зрительных нервов, колобома зрительного нерва;
- Маленькие кисти и стопы, отсутствие или значительное недоразвитие проксимальных отделов конечностей, вследствие чего кисти и стопы кажутся прикрепленными непосредственно к туловищу, уменьшение количества пальцев;
- Мраморная кожа;
- Гипоплазия сосков;
- Гипертрихоз;
- Судороги;
- Врожденные пороки внутренних органов (сердца, почек, пилоростеноз, крипторхизм) и др.
У части больных наблюдается бег по кругу, стереотипные движения руками и самоповреждения.
У всех больных отмечаются отставание в росте, в 80 % случаев — тяжёлая умственная отсталость (имбецильность), в оставшихся 20 % менее выраженные интеллектуальные нарушения; типичны рецидивирующие респираторные инфекции. Выделяют два варианта синдрома: первый (классический) с выраженной пренатальной гипоплазией, значительной задержкой физического и интеллектуального развития, грубыми пороками развития; второй — с аналогичными лицевыми и малыми скелетными аномалиями, но пограничной задержкой психомоторного развития и отсутствием грубых пороков развития.
зубоврачебный
Мелкие зубы и зубы толпились могут быть следствием малого развития челюсти у людей с ДКСТ. Гигиена зубов может быть проблематичным и усугубляется гастроэзофагеального рефлюкса, где присутствие пищеварительных кислот в полости рта гниют зубы. Ранний стоматологическая Переоценка и при переходе лечения и профилактические стратегии могут помочь преодолеть большинство проблем. Расщелины нёбо являются общими в случаях ДКСТ и это может вызвать проблемы с ушной канал и рецидивирующих инфекций уха. Если расщелина нёба отремонтированы вскоре после рождения, проблемы с едой и устной речи, которые в противном случае могли проявленной, можно предотвратить. Тем не менее, потеря слуха может варьироваться от легкой до глубокой и может измениться сверхурочно. Узкие каналы уха и трудности в проведении тщательного обследования в связи с поведением пациента, представляют вызов любому отоларинголог.
Лечение CDLS
Лечение направлено на устранение симптомов CDLS, чтобы пострадавшие могли вести более нормальную жизнь. Младенцы получают пользу от программ раннего вмешательства для улучшения мышечного тонуса, решения проблем с питанием и развития мелкой моторики. Специалисты по психическому здоровью могут помочь управлять поведенческими симптомами состояния. Детям, возможно, также понадобится обратиться к кардиологам или офтальмологам по поводу проблем с сердцем и глазами. Ожидаемая продолжительность жизни нормальная, пока у ребенка нет серьезных внутренних нарушений, таких как пороки сердца.
Каковы особенности и характеристики?
Есть много особенностей и характеристик, связанных с ДКСТ. Наиболее распространенными являются:
- Низкая масса тела при рождении – обычно ниже 2,5 кг
- Малый головы (микроцефалия)
- Задержка роста и развития – люди с ДКСТ короткие
- Черты лица – длинные ресницы, тонкие брови, которые часто соединяются, маленький вздернутый нос сужают поворачивал губы, низко расставленными ушами, волчья пасть
- Limb аномалии – укороченные, деформированные или отсутствующие конечности или цифр. Руки и ноги маленькие, мизинец часто свернувшись в, а второй и третий пальцы могут быть частично соединены
- Пороки сердца (20-30% случаев)
- Желудочно-кишечные проблемы (85% случаев), например, рефлюкс желудка, рвота, боль при приеме пищи приводит к плохой аппетит
- Задержка развития – может быть легкой или тяжелой, и может повлиять на связь
- вопросы Поведение – например, агрессия, самостимуляция, саморана (например, битье головой, рука кусаться). Это может продолжаться в циклах, и часто основной медицинской причиной.
- Другие признаки могут также произойти, например, аутистического спектра, обсессивно-компульсивное расстройство, синдрома дефицита внимания, синдром дефицита внимания с гиперактивностью. Может включать в себя, избегая контакта с глазами, ритуалистические модели поведения, и любовь к жизни.
Причины и факторы риска заболевания
До настоящего времени, этиология и патогенез этого редкого заболевания остается невыясненной до конца. Основной теорией возникновения этой болезни, считается наследственная патология, развивающаяся вследствие генетической мутации в генах.
Описаны случаи выявления этого синдрома у членов одной семьи. Наиболее вероятными факторами риска, способствующими возникновению этой патологии, являются:
- кровное родство между матерью и отцом;
- тяжелая вирусная инфекция у беременной женщины, особенно на первых месяцах зачатия ребенка, то есть во время закладки органов будущего малыша;
- побочные действия некоторых лекарственных препаратов;
- возраст беременной женщины свыше 35 лет;
- зрелые и старшие годы родителя отца будущего ребенка;
- применение физиотерапевтических процедур во время беременности;
- нарушения в эндокринной системе организма будущей матери.
Причины
Считается, что подавляющее большинство случаев вызвано спонтанными генетическими мутациями. Это может быть связано с мутациями, влияющими на когезин сложный.
-
Читайте также:

По состоянию на 2018 год было подтверждено, что с этим заболеванием связано 500 генетических мутаций; происходящий на 7 разных генах. Примерно в 30% случаев CdLS генетическая причина остается не обнаруженной. Широкая вариабельность фенотипа объясняется высокой степенью соматического мозаицизма при CdLS, а также различными генами и типами мутаций. По этой причине люди с CdLS могут иметь очень разный внешний вид, способности и связанные с ними проблемы со здоровьем.
| Имя | OMIM | Ген | Прил. % | Примечания |
|---|---|---|---|---|
| CDLS1 | НИПБЛ | 50% | Ген, ответственный за CdLS на хромосома 5 была открыта в 2004 г. совместно исследователями Детская больница Филадельфии, СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ и исследователи из Университета Ньюкасла, Великобритания. | |
| CDLS2 | SMC1A | 5% | В 2006 г. появился второй ген на Х хромосома, была обнаружена итальянскими учеными. | |
| CDLS3 | SMC3 | 1% | О третьем открытии гена было объявлено в 2007 году. хромосома 10 и был также обнаружен исследовательской группой в Филадельфии. |
Последние два гена, по-видимому, коррелируют с более легкой формой синдрома.
В 2004 г. исследователи Детская больница Филадельфии (США) и Университет Ньюкасл-апон-Тайн (Англия) идентифицировали ген (NIPBL) на хромосоме 5, который вызывает CdLS при его мутации. С тех пор были обнаружены дополнительные гены (SMC1A, SMC3 и HDAC8, RAD21), которые вызывают CdLS при изменении. В июле 2012 года был объявлен четвертый «ген CdLS» – HDAC8. HDAC8 – это ген, сцепленный с Х, то есть он расположен на Х-хромосоме. Лица с CdLS, у которых есть изменение гена в HDAC8, составляют лишь небольшую часть всех людей с CdLS. Свидетельства сцепления в хромосоме 3q26.3 неоднозначны.
Генетические изменения, связанные с CdLS, были идентифицированы в генах. НИПБЛ, SMC1A и SMC3 а также недавно идентифицированные гены RAD21 и HDAC8. Все эти генетические изменения, происходящие у пациентов с CdLS, влияют на белки, которые функционируют в когезин путь. Белки SMC1A, SMC3 и RAD21 являются структурными компонентами комплекса когезинового кольца. NIPBL участвует в загрузке когезинового кольца на хромосомы, а HDAC8 деацилирует SMC3 для облегчения его функции. Путь когезина участвует в сцеплении сестринские хроматиды в течение митоз, Ремонт ДНК, сегрегация хромосом и регулирование развития экспрессия гена. Теоретически дефекты этих функций лежат в основе некоторых функций CdLS. В частности, дефектная репарация ДНК может лежать в основе признаков преждевременного старения.
Обширный перечень симптомов
Определить наличие аномалии можно визуально сразу после рождения младенца. Ребенок появляется на свет с дефицитом веса (2/3 от общепринятой нормы). Синдром Корнелии де Ланге у пациента проявляется отличительными чертами в строении скелета, сопровождается неврологическим расстройством, отсталым умственным развитием.
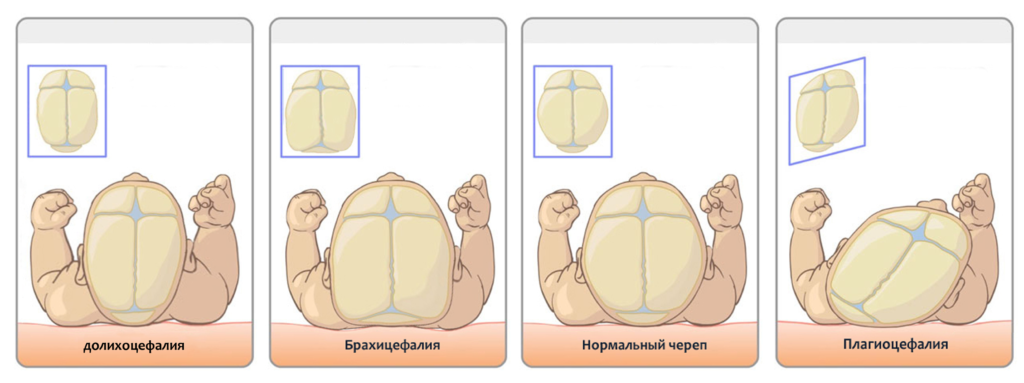
Аномалии головы
Отмечается:
- объем меньше на 15%, чем у здорового новорожденного (микроцефалия);
- ширина больше продольного размера (брахицефалия);
- верхняя (мозговая) часть уменьшена.
Дефекты лица:
- четко очерченные брови, сросшиеся (сплошная линия), негустые, недоразвитые надбровные дуги;
- ресницы длинные, пушистые, загнутые кверху, разрез глаз – по монголоидному типу;
- переносица запавшая, широкая, нос маленький, с явно выраженными ноздрями;
- нижняя челюсть не пропорциональна лицу, маленькая, квадратной формы;
- губы тонкие, с опущенными уголками;
- редкие зубы (олигодонтия) или полное их отсутствие;
- небо высокое, без перегородки (волчья пасть).
В 50% случаев выявляется повышенная волосистость тела в основном в области спины, низкая линия роста волос на лбу и затылке. Кожный покров имеет синюшный оттенок, хорошо просматривается сетка сосудов (мраморность). Последний признак не относится к основным показателям синдрома.
Недоразвитость внутренних органов
Аномалии внутренних органов являются серьезной угрозой для жизни индивида. При заболевании отмечается:
- закрытость носовых проходов (атрезия хоан), пациент дышит при помощи зонда;
- пороки со стороны сердца: дефекты в структуре сосудистой системы, перегородок, клапана, отверстие в предсердии;
- нарушения желудочно-кишечного тракта: сужение привратника, подвижность слепой кишки, грыжа на диафрагме, желудочный рефлюкс, стеноз пищевода;
- аномалии развития в мочевыводящей системе;
- недоразвитость половых органов: маленький половой член, пустая мошонка (отсутствие яичек), крипторхизм, двурогая матка;
- новообразование на почках, гидронефроз.
Также патологию сопровождают дефекты в мозговой ткани, характеризующиеся аплазией мозолистого тела и дисплазией извилин.
Костно-мышечный аппарат
При синдроме отмечается патологическое строение конечностей:
- укороченные руки по отношению к телу, отсутствие предплечий;
- неподвижность локтевого сустава (анкилоз) в результате образования хряща;
- сращивание нескольких пальцев (синдактилия), искривление пятого (клинодактилия);
- фаланги квадратной формы, приплюснуты;
- фокомелия, развитые ступни и кисти на фоне микромелии (короткие руки и ноги);
- отсутствие бугорков мышц на мизинце и большом пальце;
- образование глубоких складок на ладони;
- нижние конечности подвержены аналогичным изменениям.
Дефекты в развитии позвоночника, рост ниже нормы, соответствующей году ребенка, деформированная грудная клетка, короткая шея.
https://youtube.com/watch?v=pO88VQVjr_8
Неврологические отклонения
- косоглазие, близорукость, атрофия глазного нерва, птоз (опущение верхнего века);
- гипертонус мышц, судороги;
- нарушение в работе сегментарного аппарата, которое приводит к повышению рефлексов;
- расположение рук на уровне глаз;
- хождение по кругу;
- нарушение сна;
- частичная потеря слуха или глухота;
- склонность к самоповреждению, депрессии.
У ребенка с возрастом развивается синдром дефицита внимания, он не может длительное время заниматься одним делом, концентрация отсутствует, проявляются признаки гиперактивности, отмечается невроз навязчивых состояний.
Умственное развитие
Неврологические отклонения и аномальные изменения в головном мозге влияют на умственное развитие, способствуют проявлению имбецильности. В легкой форме патологии пациент способен к обучению, словарный запас находится в рамках бытового общения. Такие индивиды посещают детские учреждения, но при этом не обладают критическим мышлением, трудно сходятся со сверстниками, возможно проявление аутизма.
Более тяжелая форма сказывается на коммуникативных способностях. Адаптация к социуму проходит в специализированных учреждениях. Склонность к проявлению агрессии. Невозможность самостоятельного существования в незнакомой обстановке. Пациенты неспособны к самообслуживанию, требуют ухода со стороны близких людей. Тяжело поддаются обучению. С годами процесс деградации только усугубляется.
образование
Образование играет ключевую роль наряду с медицинским вопросам и имеет решающее значение в преодолении многих проблем. Учебные планы, тщательное размещение образование, где обучение может быть уникальным для человека, помогая пациентам ДКСТ минимизировать свои физические ограничения и проблемы, и помогая им развивать их самом полном объеме требует междисциплинарного подхода от речи и трудотерапии, физиотерапевтов, учителей врачей и конечно же родители и воспитатели.
Корнелии де Ланге Фонд основан в Avon Коннектикут он обеспечивает бесплатный информационный пакет по запросу и работает для поддержки семей, профессиональных и других.
Формы и симптомы болезни
Дети, рожденные с такой патологией как синдром Корнелии де Ланге, на фото выглядят определенным образом и имеют схожие черты лица, которые проявляются следующим образом:
- густой и низкий, в области лба, волосяной покров, не характерный для нормального новорожденного;
- сросшиеся брови;
- длинные и загнутые кверху ресницы;
- деформация ушных раковин;
- маленький нос с открытыми и видными носовыми ходами;
- узкая красная кайма верхней губы;
- аномально большое расстояние от верхней губы до кончика носа;
- неправильная форма черепа в виде микроцефалии (уменьшение общего объема головы) или брахицефалии, то есть увеличение горизонтального размера черепа с одновременным снижением его высоты;
- развития патологии в ротовой полости и носоглотке в виде аркообразного неба с наличием расщелины;
- мраморный рисунок кожи лица.
Кроме определенных черт лица, присущих детям с таким нарушением как синдром Корнелии де Ланге, отмечаются множественные симптомы патологии внутренних органов. Некоторые могут визуально выявляться сразу после рождения, а некоторые, в процессе дальнейшей жизни. Наиболее частыми диагностированными аномалиями внутренних органов являются:
- Патология со стороны сердечной мышцы, в виде различных пороков сердца и сосудистых нарушений. Наиболее частой аномалией развития сердечно – сосудистой системы, является дефект межжелудочковой перегородки и тетрада Фалло.
- Нарушения со стороны ЖКТ, проявляющиеся часто в виде дисфункции пищеварительной системы, вследствие чего такие дети страдают плохим аппетитом, частыми проблемами с функцией кишечника. Рвота, запоры, сменяющиеся поносами, способствуют плохому усвоению пищи и прибавлению в весе.
- Патология со стороны мочевой системы часто диагностируется в виде одиночных или множественных кистозных образований почки, аномалии развития ее. Это может быть подковообразная почка, сопровождающаяся гидронефрозом, или ее удвоение. На этом фоне имеет место развитие хронической почечной недостаточности.
- Аномалия развития половых органов у девочек, в виде двурогой матки, а у мальчиков может быть в виде крипторхизма, то есть, отсутствие яичек в мошонке.
Но наиболее тяжелыми являются последствия аномального развития центральной нервной системы. Вследствие наличия микроцефалии или неправильной формы черепа, отмечаются органические поражения ЦНС, сопровождающиеся повышением внутричерепного давления и проявляющиеся в следующем виде:
- нистагм (подергивание) при краевых отведениях глазных яблок или сходящееся косоглазие (стробизм), сопровождающиеся нарушением формы хрусталика, близорукостью или атрофией зрительного нерва;
- наличие судорожного синдрома, вследствие гипертензии головного мозга;
- парезы или параличи с нарушением функции тазовых органов;
- повышенный или пониженный тонус мышц;
- недоразвитие кистей и стоп в виде наличия четырех или трех пальцев на руках или ногах;
- отставание в психическом развитии от дебильности в легкой степени, что наблюдается в исключительных случаях, до выраженной олигофрении и имбецильности в своем большинстве;
- психомоторная расторможенность.
В клиническом течении синдрома Корнелии де Ланге, выделяются следующие его формы:
- Классический вид – когда все признаки заболевания ярко выражены. Это характерные черты лица, наличие аномалии развития внутренних органов и отставание в психическом и физическом развитии ребенка. Как правило, такие дети являются инвалидами 1 группы; требующие постоянного ухода за ними.
- Стертый вид – когда присутствуют характерные черты лица, но патология внутренних органов не является угрозой для их жизни. Нарушения функционального статуса ребенка, его психического и интеллектуального развития, выражены незначительно.
Как улучшить Средняя продолжительность жизни
Средняя продолжительность жизни у больных ДКСТ будет варьироваться в зависимости от различных факторов. Кроме правильной и своевременной диагностики, другой элемент, который может улучшить продолжительность жизни работает со специалистами
Очень важно, чтобы врачи, которые знакомы с синдромом, чтобы помочь с его руководством. Специалисты не только оказывать медицинскую помощь, но и соответствующую информацию, а также
Родители заботиться о ребенке с ДКСТ должны быть осведомлены о лучших способов идти об этом. Генетическое консультирование также необходимо для семей с ребенком, страдающим от ДКСТ. Эксперт в области ДКСТ будет направить пациента к другим специалистам, такие как генетиков, чтобы дать соответствующие указания. Родители также должны знать, как переход ребенка от педиатрической помощи в системе школьного образования и, наконец, во взрослую жизнь. Необходимо также, чтобы иметь индивидуальное лечение и управленческие возможности для конкретной семьи и пациента. Имея профессиональное руководство сделает это менее сложным.
Надзор является еще одним аспектом, который улучшает продолжительность жизни пациента ДКСТ. Ребенок страдает от психической инвалидности может иметь самоагрессивного тенденции. Сохраняя часы на такого ребенка уменьшит шансы их повредить себя или других лиц; и, следовательно, улучшить их продолжительность жизни. Правильный надзор будет также сделать его легче заметить проблемы со здоровьем, которые могут оказаться смертельными. Получение специализированной помощи является одним из вариантов, которые родители считают, когда уход за ребенком с ДКСТ. Мониторинг роста и принимая ребенка для тестирования также поможет с управлением синдрома и повышения продолжительности жизни.
Средняя продолжительность жизни человека с ДКСТ будет зависеть не только от управления состоянием, но препараты, изготовленные на разных этапах жизни. Например, пациент может узнать навыки, такие как мойка, чтобы дать им возможность пользоваться какой-то степени независимости. Пациент с связи и речи impartment могут получить лечение, чтобы помочь им общаться с помощью различных средств, таких как язык жестов. Такие навыки сделает его менее сложным для страдалец ДКСТ, чтобы жить дольше, тем самым, увеличивая продолжительность жизни.
Причины синдрома
Современная медицина пока только выдвинула две теории развития болезни, причем не одна из них полностью не доказана. На первом месте стоит тема генетических аномалий, связанных с передачей гена, отвечающего за болезнь. Хотя при подробном изучении заболевших и их родственников так и не удалось выявить четкого раздробления хромосом.
Можно отметить только опыт изучения нескольких семей, в которых дети были рождены с синдромом Ланге. В этих семьях регистрировались люди, страдающие генетическими аномалиями. У некоторых было недоразвитие конечностей, некоторые страдали от невыясненных аномалий со стороны количества пальцев на руках и ногах. А также у кого-то из родственников прослеживалось явление нанизма, то есть карликовости и недоразвития. Нанизм, проявляющейся умственной отсталостью почти всегда регистрируется у самих пациентов с синдромом Ланге.
В последнее время все-таки ученые не отрицают еще один путь зарождения болезни – это аномалии в период беременности или люди, попадающие в группу фактора риска. Было изучено несколько случаев синдрома, когда мать в период беременности употребляла большое количество химиотерапевтических препаратов, подвергалась физиотерапевтическим процедурам. То есть нельзя исключать теорию токсического воздействия на плод в первый триместр беременности.
У некоторых детей прослеживалась взаимосвязь с заболеваниями и состоянием матери до и в период беременности. Так, к факторам риска можно отнести эндокринные патологии, инфекционные заболевания, радиоактивное облучение. Еще один фактор риска относится не только к матери, но и к отцу – это возраст человека. Дети с аномалиями развития появлялись у родителей, которые решили забеременеть в возрасте после 35 лет либо были кровными родственниками.
Костно-мышечные симптомы
- Задержка костного возраста : пренатальные и постнатальные аномалии роста могут вызвать значительную задержку созревания костей.
- Гипоплазия: у значительной части пораженных наблюдается асимметричное развитие конечностей и членов тела. Обычно руки и ноги меньше обычных.
- Синдактилия: при этом синдроме часто встречается сращение кожи или костной ткани некоторых пальцев рук.
- Брахиклинодактилия: пятый палец руки обычно изогнут и искривлен.
- Oligodactyly: отсутствие одного или нескольких пальцев рук или ног является еще одним из костно – мышечной функции , которые могут быть идентифицированы.
- Мышечная гипотония: тонус мышечной структуры обычно вялый или ненормально сниженный.
Что такое синдром Корнелии де Ланге
Пациенты с редкой патологией рождаются, не только имея множественные отклонения от нормы. Специалисты диагностируют симптомы умственной отсталости после появления малыша на свет.
Родители маленьких пациентов утверждают, что дети находятся в постоянном раздражении. Они не умеют пользоваться туалетом и не пытаются учиться этому, совершают различные действия бессмысленного характера.
Ученые определили 2 формы заболевания:
- Классический синдром. Патологические процессы сопровождаются ярко выраженными признаками. Дети рождаются со специфической внешностью, у них диагностируют многочисленные пороки. В сложных ситуациях у больных развивается умственная отсталость тяжелой формы. Обучение таких детей невозможно.
- Стертый вид болезни. У ребенка появляются дефекты не только на лице, но и по всему телу. Внутренние органы находятся в безопасности, патологические процессы на них не распространяются. Жизни малыша ничего не угрожает. Присутствуют слабые нарушения, связанные с моторикой, психикой и интеллектом.
Терапию в любом случае подбирает врач
Важно постоянно следить за состоянием больного и придерживаться всех рекомендаций врача
Фото заболевания:

Как диагностировать Cornelia De Lange синдром?
Из-за большого разнообразия характеристик и особенностей, ДКСТ обычно клинически диагностирована специалистами. Это будет включать в себя взятие истории болезни, проведение медицинского осмотра и организации генетического тестирования (особенно для мутаций в гене NIPBL). Хотя генетические мутации выявляется почти в 60% случаев, иногда тесты не показывают их. Тем не менее, это, как полагают, потому что есть другие гены, участвующие в ДКСТ, которые еще не были изолированы, так как только они были определены и подтверждены, генетическое тестирование может включать их.
Внутриутробное диагноз ДКСТ трудно, из-за ряда факторов, участвующих. Если ребенок рождается с ДКСТ, рекомендуется родителям, что высокой четкости ультра-звук сканирование осуществляется в течение 18-й недели любой последующей беременности. Это может сосредоточиться на наблюдаемых особенностей, таких как аномалии конечностей и пороков сердца, но не может обеспечить окончательный дородовой диагностики.




